About
• Objective
• Applications
• Script
• Shapes
|
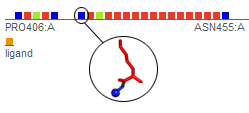
The Pocket Creator ObjectiveProtein receptors are often studied in a reduced form that concentrates on the ligand binding site and includes only residues that are at some distance from the ligand. Computing all the interaction energies between the atoms of a ligand and a full protein receptor is time consuming and most computations of protein-ligand interactions are truncated by some cutoff distance or by some reduction of the protein structure. The large number of non-bonding interactions may thus focus on the most crucial ones. Cutoff distances are especially common if interactions have to be computed frequently, such as in molecular dynamics or in virtual docking. In docking of large databases, residues that align the ligand binding site or are close neighbors have a larger contribution to binding by electrostatic or Van der Waals interactions as well as by their surface interaction with the ligand, and it is unnecessary to include the full protein. However, truncating a protein to form a pocket around a ligand frequently creates partial chains and requires amendments to overcome structural problems due to spurious chain terminations and to short breaks in chains and in secondary structures (see images below). Our script called Pocket Creator, tries to tackle that problem and creates binding pockets with minimal interference to the overall structure by adding small gaps and breaks to the cutoff structure (see script details below). With this web site, we provide the scientific community a web interface to the Pocket Creator script which saves and displays binding pockets that are useful for further research and computations.
The Pocket Creator Applications
The Pocket Creator Script
The Pocket Shapes
|